Phenoxome: ranking pathogenic variants provided phenotypic context
A comprehensive computational framework to prioritize genetic variants based upon the likelihood of the variant being pathogenic given the phenotype of the patient. 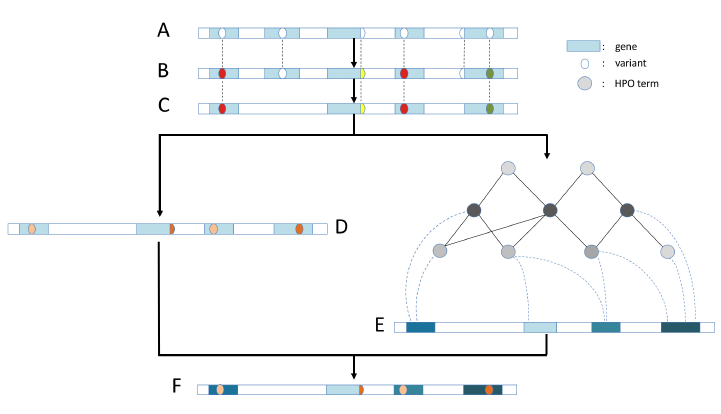

A comprehensive computational framework to prioritize genetic variants based upon the likelihood of the variant being pathogenic given the phenotype of the patient.
Identifying potential drug repurposing candidates by community detection on heterogeneous networks.
A computational classifier to distinguish sequencing artifacts from bona fide variants from next-generation sequencing data in pediatric tumors.
A computational workbench to prioritize miRNAs provided disease context represented by messengerRNA targets.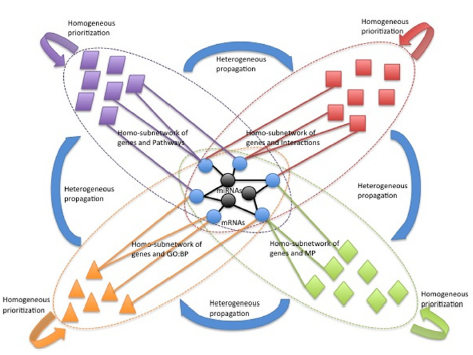
Published in Journal 1, 2009
This paper is about the number 1. The number 2 is left for future work.
Recommended citation: Your Name, You. (2009). "Paper Title Number 1." Journal 1. 1(1). http://academicpages.github.io/files/paper1.pdf
Published in Journal 1, 2010
This paper is about the number 2. The number 3 is left for future work.
Recommended citation: Your Name, You. (2010). "Paper Title Number 2." Journal 1. 1(2). http://academicpages.github.io/files/paper2.pdf
Published in Journal 1, 2015
This paper is about the number 3. The number 4 is left for future work.
Recommended citation: Your Name, You. (2015). "Paper Title Number 3." Journal 1. 1(3). http://academicpages.github.io/files/paper3.pdf
Published:
We present a computational method to identify putative drug repurposing candidates through network clustering. A weighted drug-disease network was compiled using known drug-target and disease-gene associations. The disease-drug pairs were assembled in communities detected in the heterogeneous network. Presented at ICIBM 2013. Read more
Published:
In this paper we present a novel framework capable of finding statistically significant biclusters on ENCODE ChIP sequencing datasets. Our goal is to discover low biclusters with low variance in terms of gene expression, which in theory shold point to coherent functional modules. Presented at ICDMW 2013. Read more
Published:
Phenoxome is a computational application that improves the efficiency of the interpretation of WES/WGS data by providing a prioritized list of genes coupled with variant analyses from presenting patient phenotypes. We expect more intelligent computational models will be developed to increase accuracy in variant classification and further reduce the processing time. Presented at AMP Annual Meeting 2015. Read more
Published:
We presented a computational classifier to identify true positive SNVs from tumor sequencing. The classifier demonstrated high sensitivity/specificity, and utility on a wide range of tumor samples. Overall, 96% of the SNVs detected will receive a definite label and thus be exempt from manual review. Implementing the model can greatly reduce the hands-on time and hence improve the efficiency without compromising the quality of the clinical tests. Presented at AMP Global Congress 2019. Read more
Undergraduate course, University 1, Department, 2014
This is a description of a teaching experience. You can use markdown like any other post.
Workshop, University 1, Department, 2015
This is a description of a teaching experience. You can use markdown like any other post.